Our SoluTions
Accelerate Your Discovery Programs
Find promising candidates rapidly
Instant access to world-class AI expertise and infrastructure
Save costs by skipping in-house teams
ISO 27001 certified company


End-to-End Discovery Projects
Drug Discovery Partnerships
We partner with you to to undertake a drug discovery project. We bring technology and computation, you bring the specific know-how.
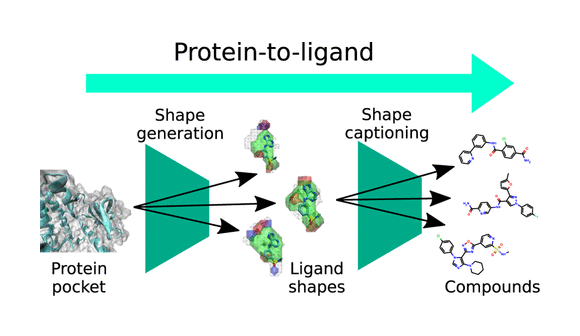
Generative AI for hit discovery
Binding free energy calculations for hit-to-lead
ADME predictive models for lead optimization

AI and simulations platform services
PlayMolecule Services
We work together with your team, delivering target-specific data using PlayMolecule, our AI and simulations platform, on our computing infrastructure.
Answering specific quesions
Discovery Services
We deliver insightful data that answers very specific questions in a limited time.
Target conformaitonal space (active/inactive conformation)
Druggable binding pockets (cryptic and allosteric sites)
Gigascale virtual screening
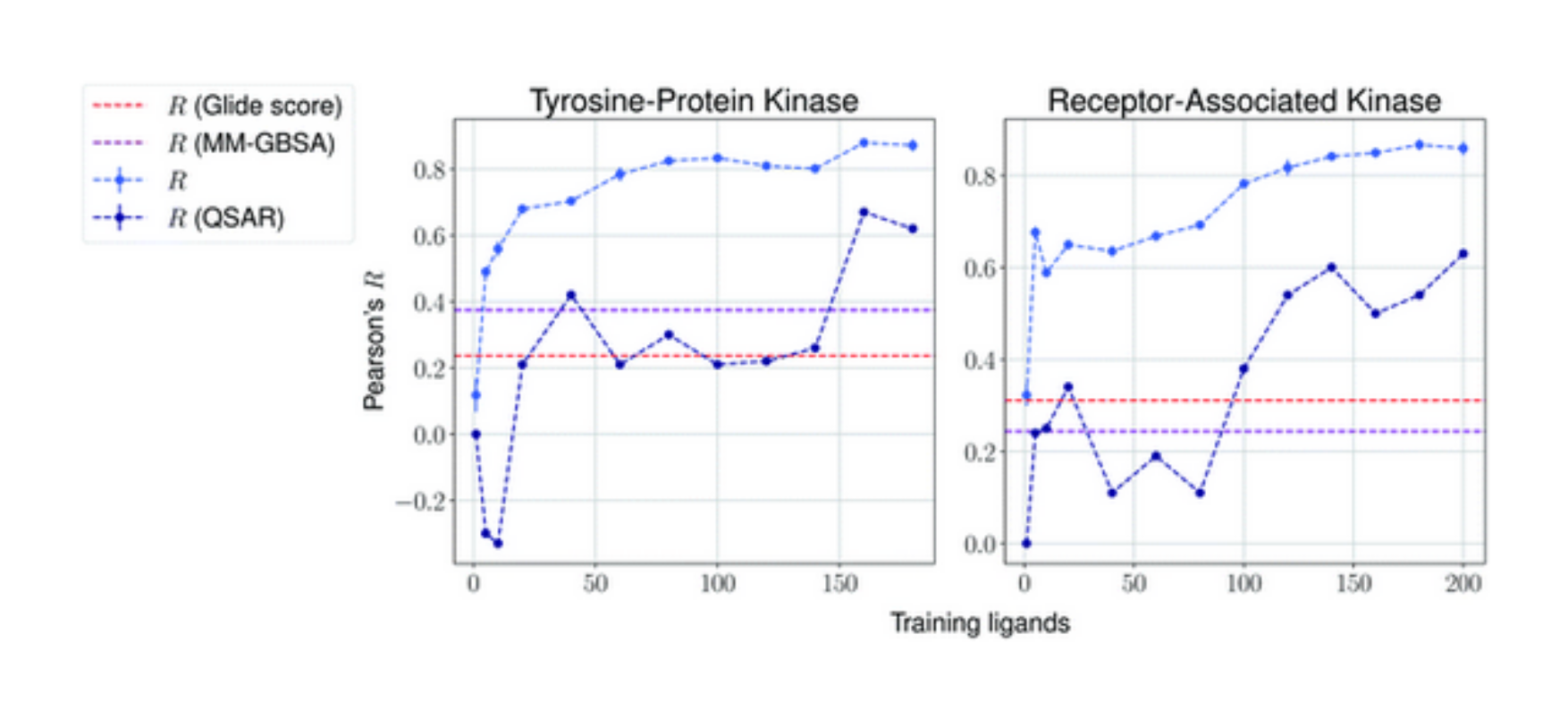
Case studies