From Drug Discovery to Drug Engineering
AI and quantum chemistry solutions for early-stage drug discovery
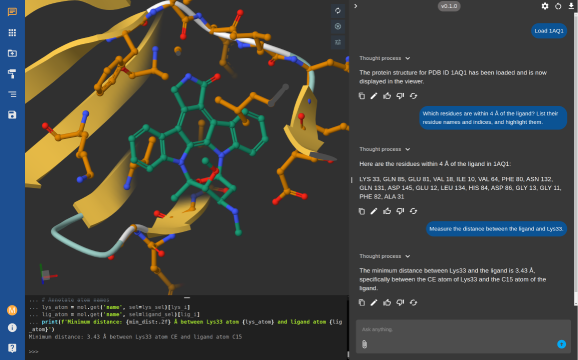
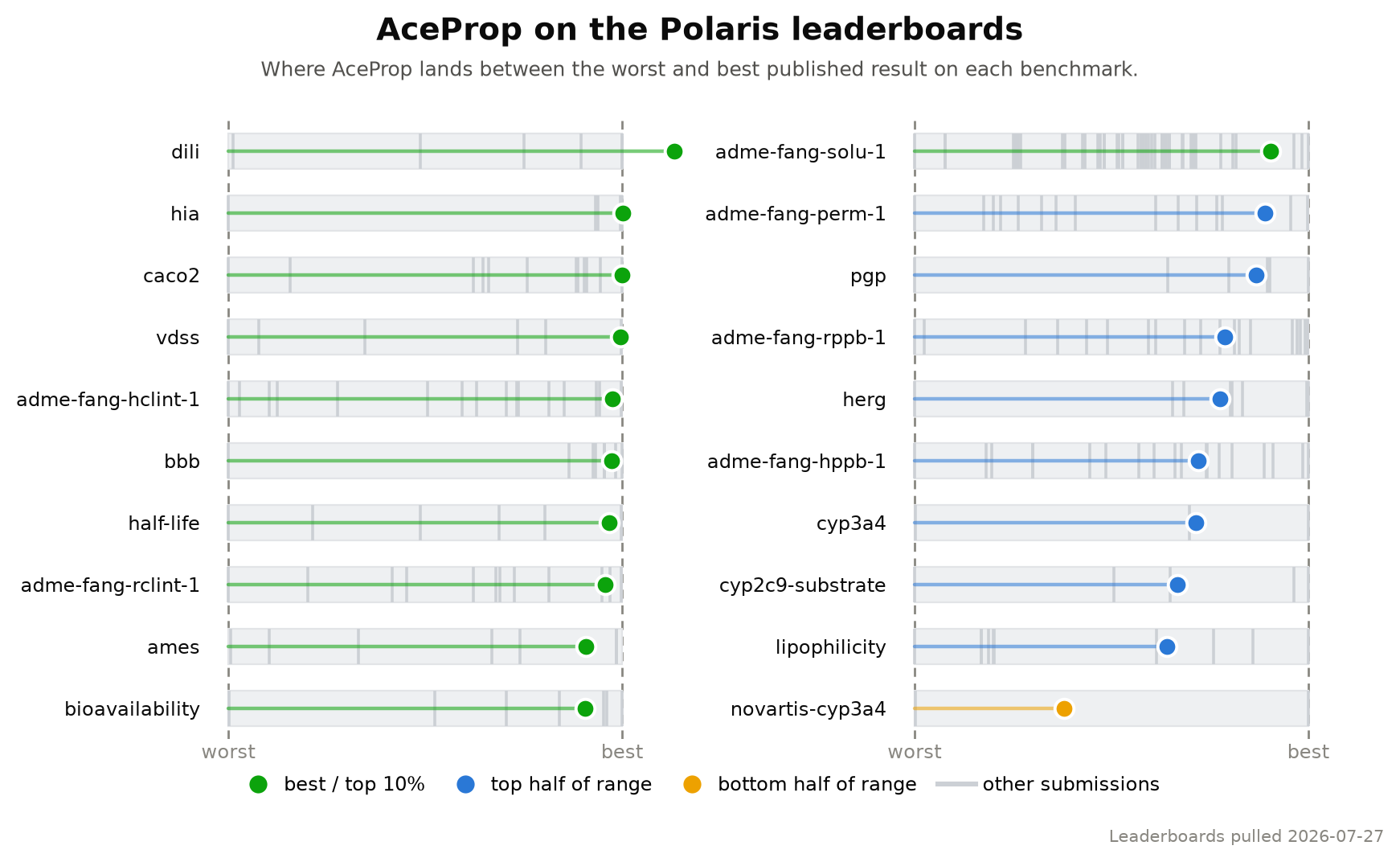
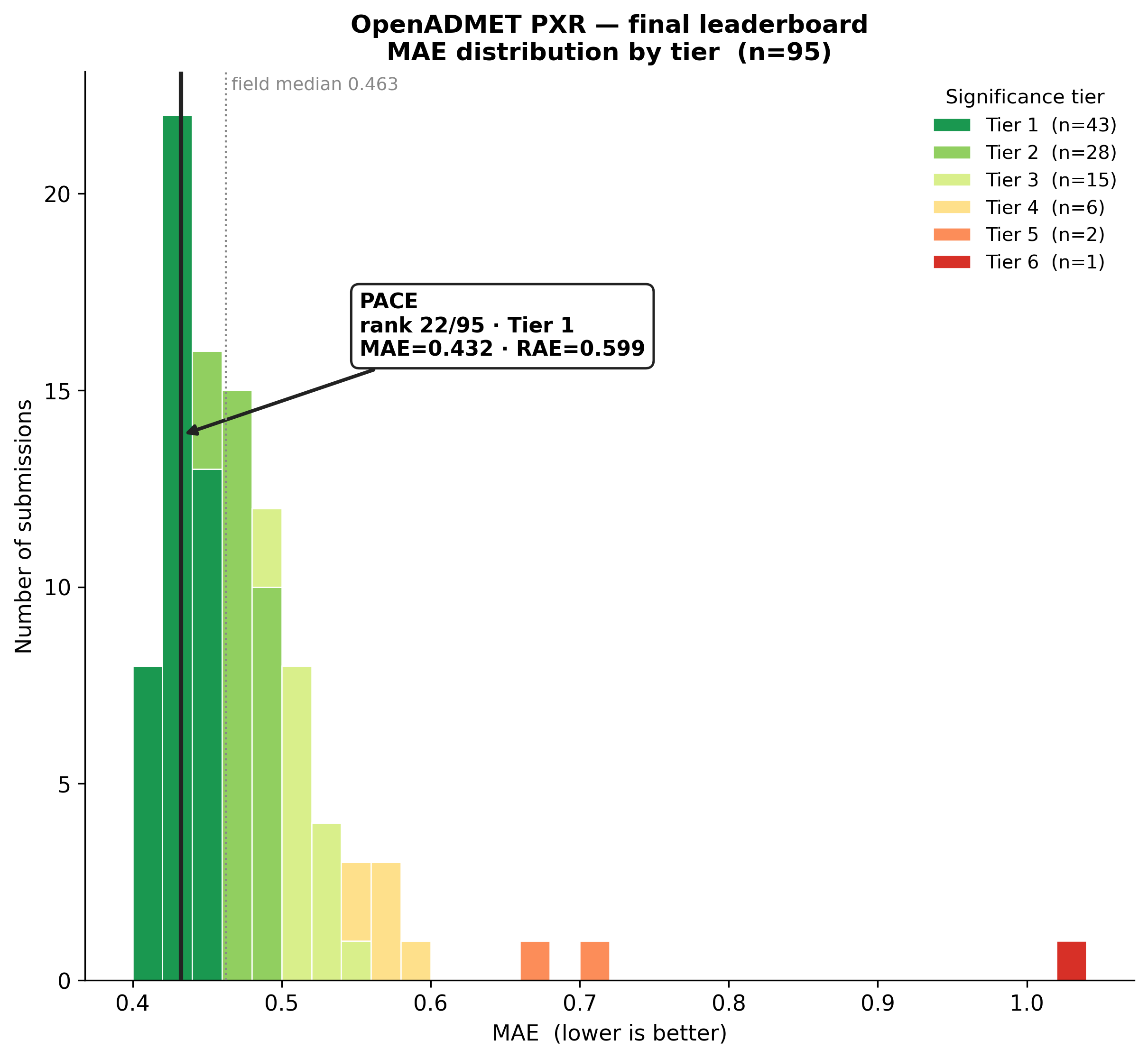
Playmolecule ai
Acellera's Enterprise AI Co-scientist
Securely connect your proprietary datasets and internal libraries to our AI platform.
Acellera Therapeutics
Innovation Leaders
Backed by years of technological and scientific innovation, our team of drug hunters, machine learning researchers, and software engineers are coming together to bring new therapies for patients through computation and AI.


Our Platform
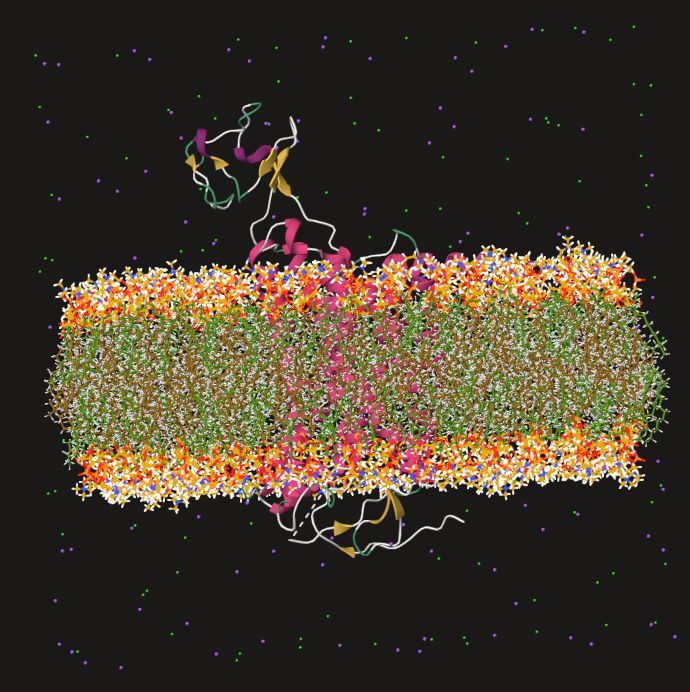
Meet QuantumBind®
Our platform for small-molecule discovery and potency optimization that combines the accuracy of quantum chemistry and the speed of AI/ML.
What We Offer
Higher Success Rates
We enhance your small-molecule discovery capabilities by shortening the time to identify lead molecules. With our technology and expertise, we can take your programs from target to a prioritized list of lead candidates within several weeks.


Case studies
Demonstrable Experience
We worked with dozens of top pharma and biotech companies worldwide to apply our technology to the toughest scientific and technological challenges in drug discovery.
NEWS & UPDATES