Stop Searching, Start Asking: Introducing "Speak to a Protein"
How long does it take you to really understand a new protein target?
For most researchers, the process is a slow, painstaking effort. It involves weeks of digging through PubMed , cross-referencing PDB structures , querying databases like UniProt and ChEMBL , and trying to visualize it all with specialized software. This "high friction" workflow requires deep computational skills and slows down the pace of discovery.
What if you could just... talk to it?
At Acellera, we're excited to introduce Speak to a Protein, a new capability that transforms protein analysis into an interactive, multimodal dialogue with an expert AI co-scientist.
This system, which we recently published on arXiv, doesn't just chat about science—it does science. It seamlessly couples natural language, 3D visualization, and real-time code execution to give you evidence, not just answers.
How an AI Co-Scientist Works
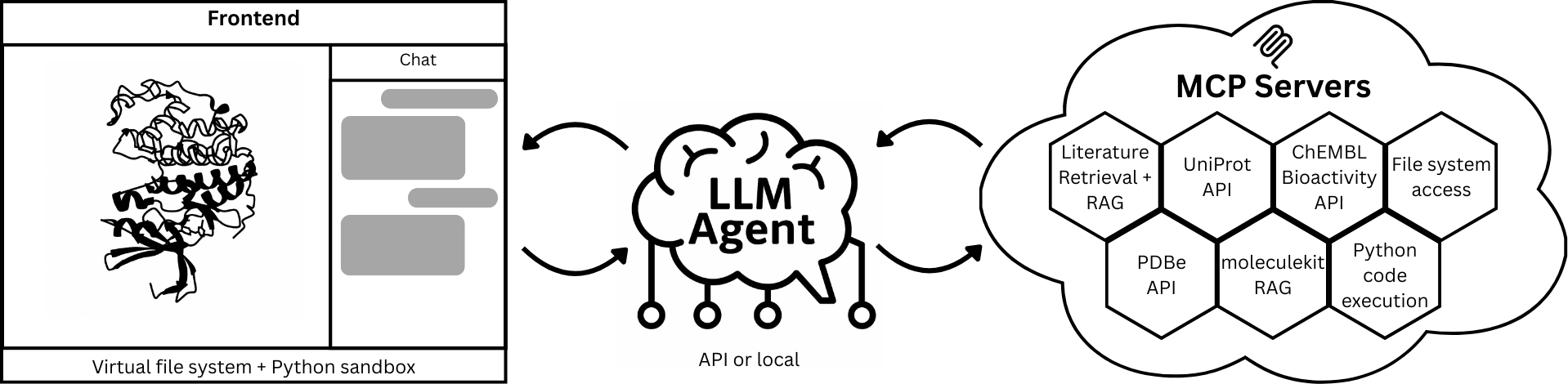
The core problem with protein analysis is that data is fragmented. Literature, 3D structures, and bioactivity data all live in different places. Speak to a Protein acts as a central hub, or "LLM Agent," that connects all these sources.
When you ask a question, the AI co-scientist:
- Retrieves & Synthesizes Data: It autonomously queries literature, structural (PDB), and biochemical (UniProt, ChEMBL) databases.
- Generates & Runs Code: It writes and executes Python code on the fly in a secure sandbox to perform calculations, filter data, or generate plots.
- Grounds Answers in 3D: It interacts with a live 3D molecular viewer to highlight residues, measure distances, or annotate binding pockets, so you can see what it's talking about.
A Quick Walkthrough: From Question to Evidence
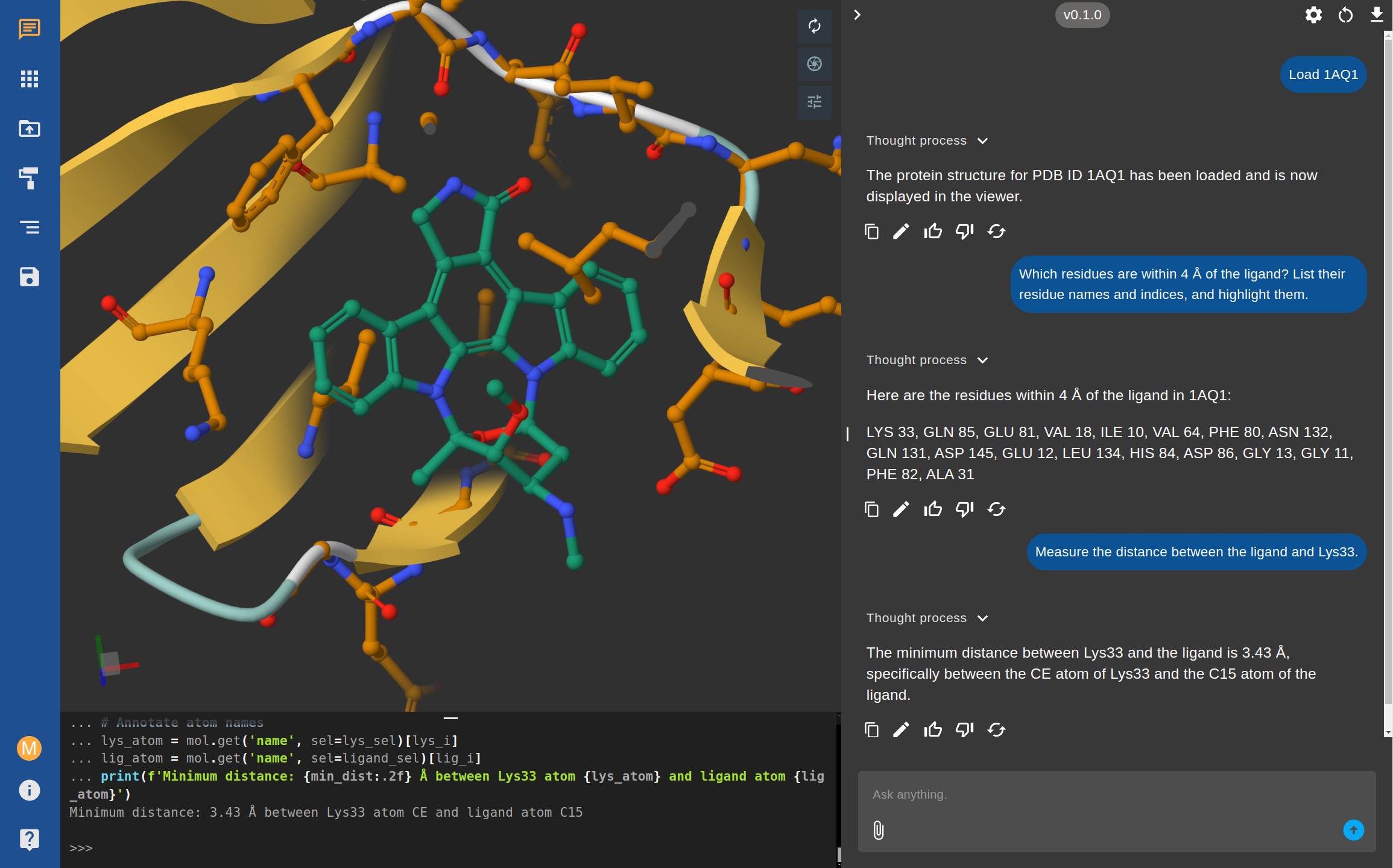
Let's say you're exploring Cyclin-dependent kinase 2 (CDK2), using the structure 1AQ1.
You can start a simple conversation:
- You: "Which residues are within 4 Å of the ligand?"
- AI: The system instantly identifies the residues (LYS 33, GLN 85, GLU 81, etc.) and, more importantly, highlights them in the 3D viewer.
- You: "Great. Now measure the distance between the ligand and Lys33."
- AI: The AI generates and runs a snippet of Python code to perform the calculation. It responds: "The minimum distance between Lys33 and the ligand is 3.43 Å, specifically between the CE atom of Lys33 and the C15 atom of the ligand."
All of this happens in seconds. You've gone from a question to a precise, verifiable piece of evidence without writing a single line of code or opening another program.

A Full Drug Discovery Workflow in Minutes
Speak to a Protein isn't just for simple lookups. It can automate entire discovery workflows that would normally take hours or days.
1. Mining Bioactivity Data
Imagine you're investigating the Dopamine D3 receptor (D3R). You can ask:
"Give me all the known inhibitors for D3R and their affinities. Load the resulting table."
The AI co-scientist understands this complex request. It identifies the UniProt entry for D3R, queries the ChEMBL database for all functional assays, retrieves the data, and presents it as a fully interactive table right in the viewer, complete with chemical structures and potency metrics (IC50, Ki, etc.).
.png)
2. Large-Scale Structure-Activity Analysis (SAR)
In our paper, we demonstrated a complete workflow on CDK2. We asked the AI to:
- Find all 462 associated PDB structures.
- Identify which of them had co-crystallized ligands relevant for drug discovery (it found 479 unique pairs).
- Cross-reference these ligands with ChEMBL to find bioactivity data (132 had it).
- Filter, clean, and deduplicate this dataset to find the top 20 most potent complexes.
- Load and structurally align all 20 of those structures in the 3D viewer.
The result? In just a few minutes, we had a fully aligned ensemble of the most potent CDK2 inhibitors, ready for immediate visual analysis of their binding modes. The system can even generate a publication-ready summary report of its findings.
Try It Today, And See What's Next
Speak to a Protein is built on our PlayMolecule platform and is freely accessible for you to try right now:
We believe this tool significantly lowers the barrier to advanced structural analysis and will accelerate hypothesis generation for researchers everywhere.
But what you see online is just the beginning.
At Acellera, we have integrated this AI co-scientist with our entire internal suite of experimentally validated computational tools, including advanced molecular dynamics (MD) simulations, binding affinity predictors, and more.
This creates a "super-powerful" co-scientist that can not only retrieve and analyze existing data but also generate new data and make novel predictions. This internal system can help guide complex drug discovery campaigns, suggest new inhibitor designs, and explain why one compound is better than another.
While this advanced version isn't publicly available due to the immense computational resources it requires, we are offering live demos to interested research teams and partners.
If you're interested in seeing the future of AI-driven drug discovery, contact us to schedule a demo.
We're incredibly excited to see what new discoveries will be made when anyone can simply... speak to a protein.