AceFF™-2: Bridging the Gap Between Speed and Accuracy in Drug Discovery
For decades, computational chemists have faced a frustrating trade-off: you could use classical Molecular Mechanics (MM) force fields (like GAFF or AMBER) to get results quickly but with limited accuracy, or you could use Quantum Mechanics (QM/DFT) to get precise results at a computational cost that makes routine simulation impossible.
Machine Learning Interatomic Potentials (MLIPs) promised to fix this, but early iterations had limits, often struggling with charged molecules or lacking support for the diverse elements found in real drug candidates.
Enter AceFF™-2.
In our latest paper [1] (https://arxiv.org/abs/2601.00581 ), we introduce AceFF™-2, a next-generation neural network potential designed specifically for small drug-like molecules. It is built to hit the sweet spot: delivering near-DFT accuracy at speeds fast enough for high-throughput screening and molecular dynamics (MD).
Key Takeaways
- Broad Coverage: Supports all essential medicinal chemistry elements (H, B, C, N, O, F, Si, P, S, Cl, Br, I) and explicitly handles charged states.
- New Architecture: Built on TensorNet2, which introduces "neutral charge equilibration" to better model long-range Coulomb interactions.
- The Model: It is significantly more accurate than fast models like ANI-2x, but orders of magnitude faster than heavy models like OrbMol.
- Production Ready: Compatible with ACEMD/OpenMM and CUDA graphs for low-latency simulations.
Why TensorNet2 Changes the Game
The predecessor, AceFF™-1.0, was a strong start, but like the popular ANI-2x model, it struggled when extrapolating to larger, charged molecules.
AceFF™-2 solves this by upgrading the architecture to TensorNet2. We incorporated a physics-informed approach similar to AIMNet2, where the model predicts partial charges and performs a neutral charge equilibration. This allows the model to calculate a Coulomb energy term alongside the standard neural network energy.
The result? A model that understands the electrostatics of complex, charged drug molecules without exploding computational costs. The parameter increase is only ~28%, with a speed decrease of less than 10% compared to the previous version.
Proven Accuracy: Putting AceFF™-2 to the Test
We validated AceFF™-2 against rigorous benchmarks, including complex torsion scans, strained conformers, and large ligand datasets.
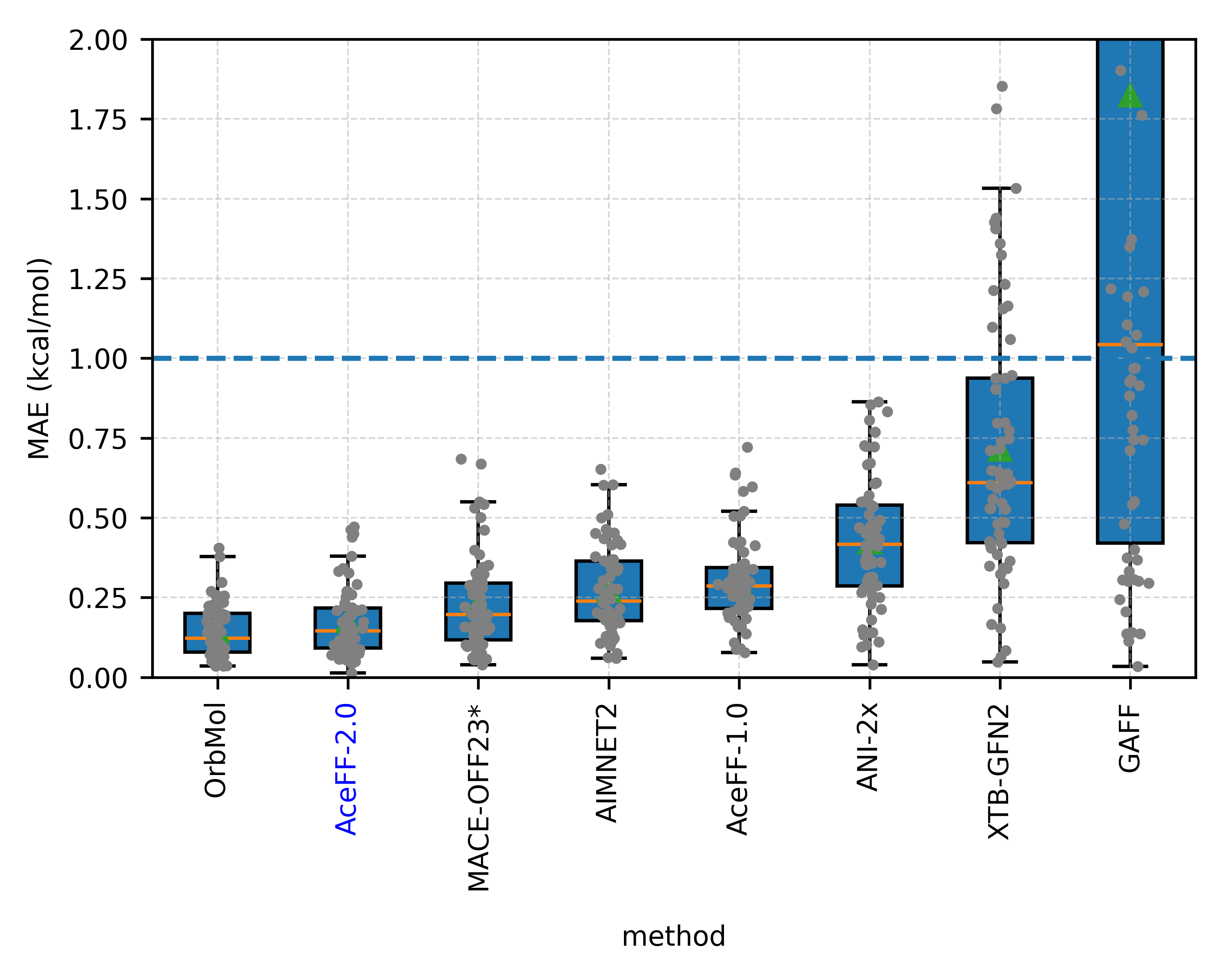
1. Torsion Scans
In the Sellers et al. torsion scan benchmark, AceFF™-2 proved to be a close second in accuracy only to OrbMol (a much heavier, slower model), while significantly outperforming ANI-2x and traditional force fields like GAFF.
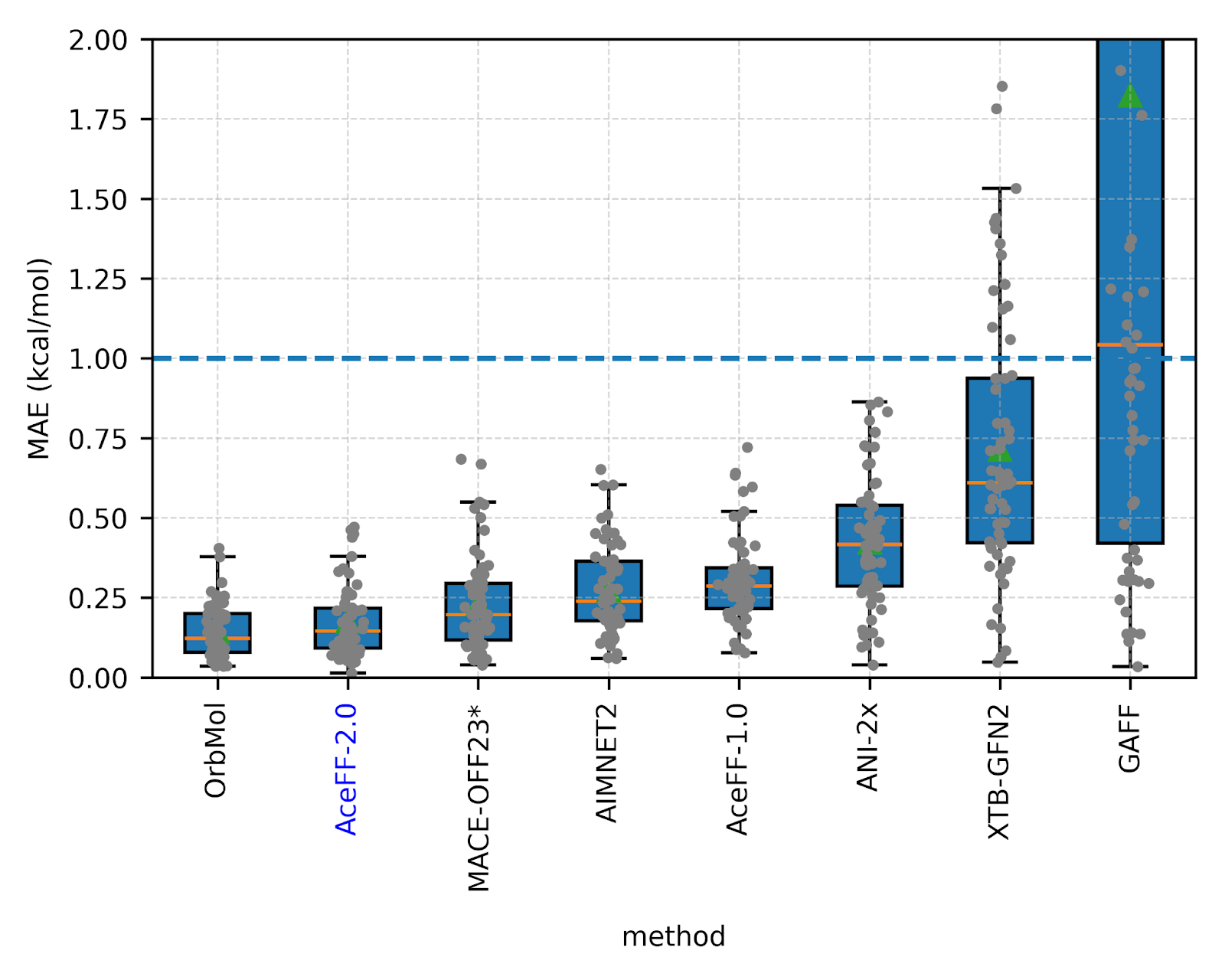
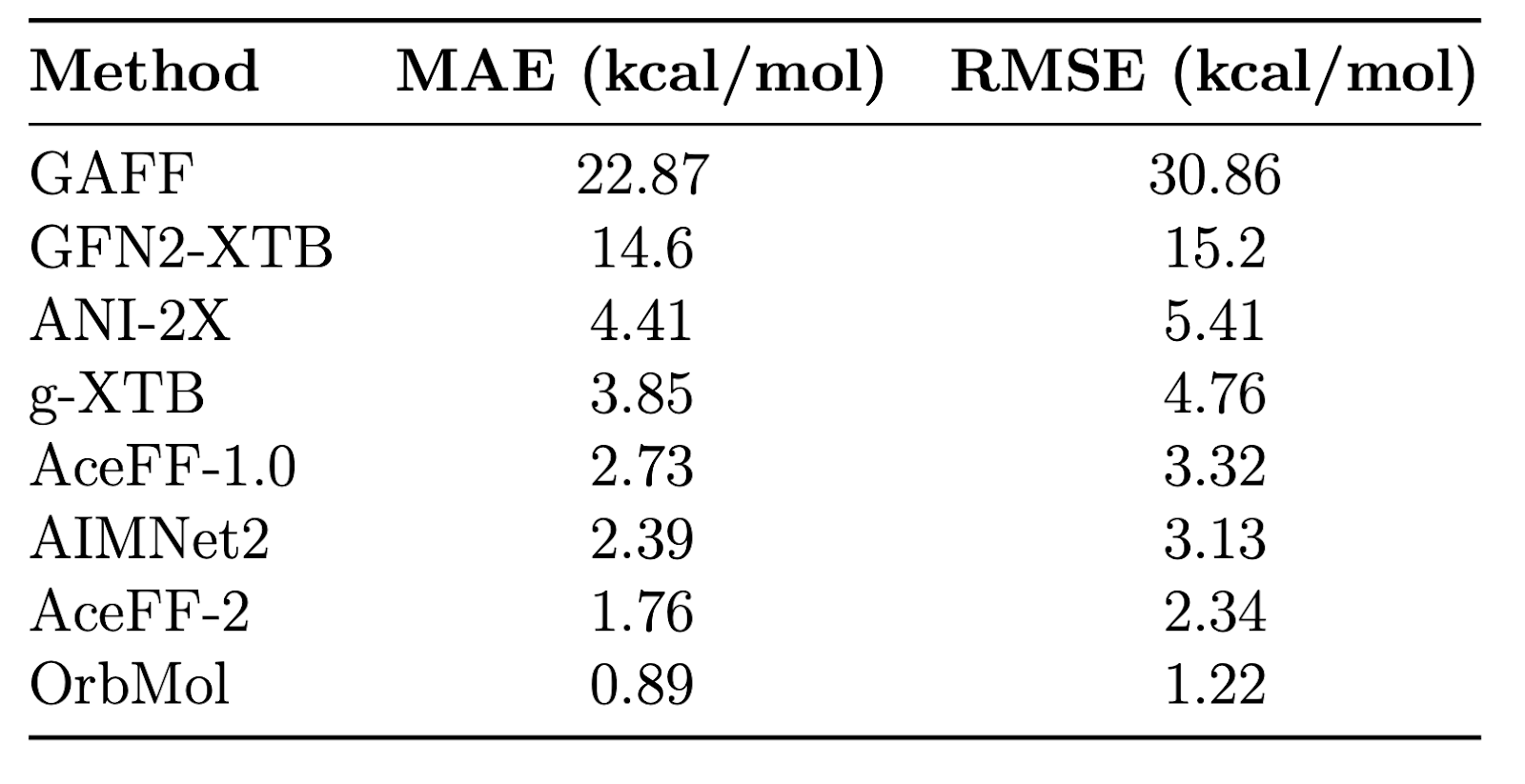
2. Handling Strain (Wiggle150)
Modeling high-energy, strained conformations is critical for understanding transition states. On the Wiggle150 benchmark, AceFF™-2 achieved a MAE of 1.76 kcal/mol, improving on AIMNet2 (2.39 kcal/mol) and the semi-empirical g-XTB method (3.85 kcal/mol).
3. Generalization (Schrödinger Ligands)
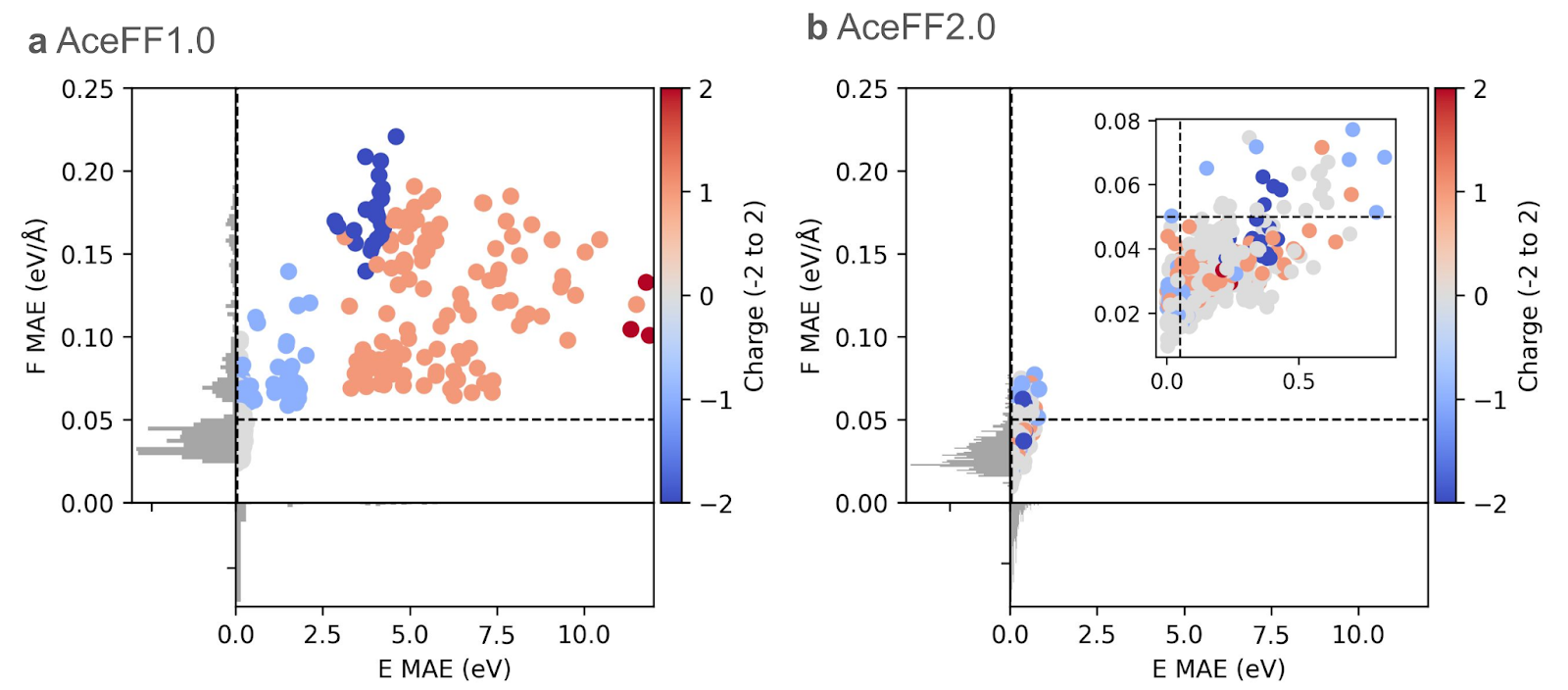
We tested the model on 650 conformers of large ligands from [5] (30+ atoms) that it had never seen before. While previous models failed on charged variants, AceFF™-2 maintained low errors, proving it can generalize well beyond its training set.
4. Smooth potential energy surface
Stability in molecular dynamics simulations hinges on the quality of the potential energy surface. To evaluate our model’s smoothness, we performed a potential energy scan by systematically compressing and extending the C-C bond in ethane. AceFF™-2 closely matches the DFT potential energy.
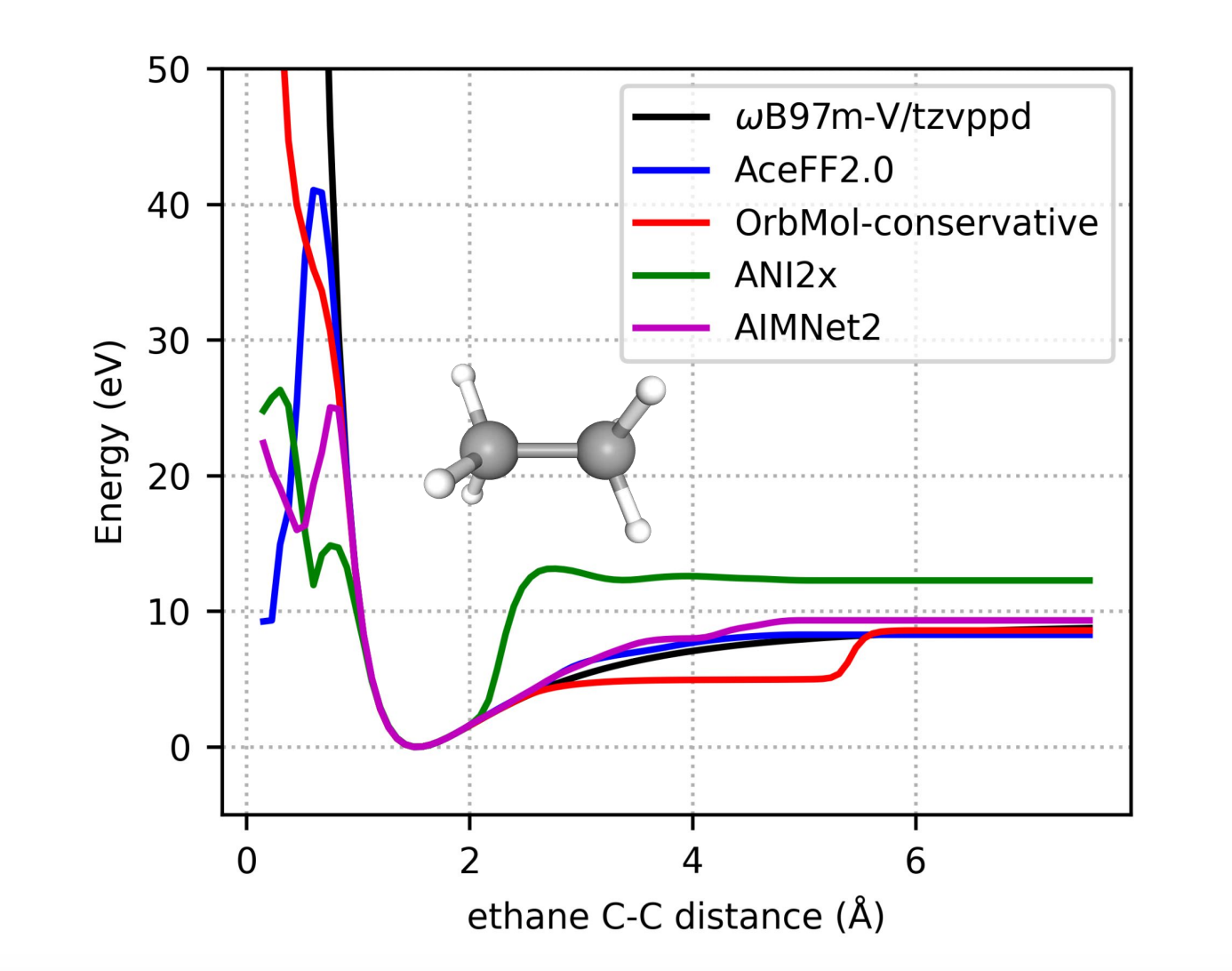
Speed That Scales
We optimized AceFF™-2 for inference speed. By utilizing CUDA graphs, AceFF™-2 achieves low latency even for small systems (under 300 atoms), a regime where other models often bottleneck.
For mixed ML/MM simulations, where the ligand is modeled with MLIP and the protein with a classical force field, AceFF™-2 is stable and fast. In our tests on an RTX4090, we achieved 36.7 ns/day with a 1fs timestep. This makes it a viable tool for actual production-level Free Energy Perturbation (FEP) workflows.
Easy Integration
AceFF™-2 is built for seamless integration into existing computational chemistry pipelines. You can easily use it through the widely adopted Atomic Simulation Environment (ASE) interface, deploy it directly within high-performance molecular dynamics simulations using ACEMD and OpenMM, or access its core functionality as a pure PyTorch model.
For example, here is the code for a single point calculation in ASE:
It is available as an ML potential in OpenMM-ML:

Get Started
AceFF™-2 is open-source (Apache 2.0) and available now. We believe it represents the most practical balance of speed and accuracy currently available for drug discovery.
- Download the Model: HuggingFace
- Tutorials & examples:
- Notebooks that can run in Colab https://github.com/Acellera/aceff_examples
- If you prefer python scripts look in the torchmd-net examples folder
- Code: https://github.com/torchmd/torchmd-net
References
[1] Stephen E. Farr, Stefan Doerr, Antonio Mirarchi, Francesc Sabanes Zariquiey, Gianni De Fabritiis, AceFF: A State-of-the-Art Machine Learning Potential for Small Molecules, https://arxiv.org/abs/2601.00581 (2026)
[2] Francesc Sabanés Zariquiey, Stephen E. Farr, Stefan Doerr, Gianni De Fabritiis, QuantumBind-RBFE: Accurate Relative Binding Free Energy Calculations Using Neural Network Potentials, https://arxiv.org/abs/2501.01811 (2025).
[3] Joseph Gair, Corin Wagen, Rebecca Brew, Ian Nelson, Meruyert Binayeva, Amlan Nayak, Wyatt SimmonsWiggle150: Benchmarking Density Functionals And Neural Network Potentials On Highly Strained Conformers. https://chemrxiv.org/engage/chemrxiv/ article-details/67f5035c6dde43c908ee4655 (2025)
[4] Sellers, B. D.; James, N. C.; Gobbi, A. A comparison of quantum and molecular mechanical methods to estimate strain energy in druglike fragments. Journal of chemical information and modeling 2017, 57, 1265–1275.
[5] https://github.com/schrodinger/public_binding_free_energy_benchmark